

Epilepsy Insights

Top 5 insights from the American Epilepsy Society meeting (2021)
The American Epilepsy Society (AES) meeting is the largest epilepsy meeting of the year, and because it takes place every month of December it also serves as an annual review on the understanding and treatment of epilepsies. These are my top 5 insights from the American Epilepsy Society 2021 meeting.
The American Epilepsy Society (AES) meeting is the largest epilepsy meeting of the year, and because it takes place every month of December it also serves as an annual review on the understanding and treatment of epilepsies. I look forward every year to the first week of December for this reason.
I look for therapies for rare genetic epilepsies, and in the recent years this area has exploded to take over much of the AES meeting. Because of that, I will focus this update on the rare genetic epilepsies, or Developmental and Epileptic Encephalopathies (DEEs). There is a lot more that is presented at the AES meeting and that I will not cover. These are my top 5 insights from the American Epilepsy Society 2021 meeting.
1. Natural History Studies are tremendously important
When companies develop treatments for seizures as a symptom, it is straightforward to run trials: just count seizures. But when companies start developing treatments that treat the cause of that epilepsy, and in particular when we are talking about complex neurodevelopmental syndromes with epilepsy, then clinical trials are more complex than just counting seizures, and need more solid data on the usual symptoms and severity that those patients have so that we can know if there is efficacy. This is where Natural History Studies (a type of observational study) specifically designed to inform on clinical trial design come into play. And we are seeing a big number of these.
Several of these ongoing studies were presented at the AES meeting. The BUTTERFLY study by Stoke Therapeutics is an observational study to characterize clinical scales to measure the non-seizure aspects of Dravet syndrome, and has already shown promise with several scales that could be used in interventional clinical trials. The ENVISION study by Encoded Therapeutics, has also helped validate scales and determine the baseline characteristics on non-seizure outcomes in young children with Dravet syndrome. Both companies are developing treatments to rescue SCN1A gene expression in Dravet syndrome, so these observational studies are designed to make those treatment trials possible, and both observational studies are already producing very useful data.
We also saw example of more proactive observational studies that are already ongoing even before companies are ready for trials, like an SCN8A Natural History Study resulting from a partnership between hospitals and patient groups, and a consortium working on STXBP1 Natural History. While these studies are simpler than the company-run Dravet syndrome studies, and don’t validate scales, they help us understand the collection of symptoms and their severity in these syndromes, and guide the design of the clinical-trial enabling studies. It is also remarkable that both studies include data from more than 500 patients from each syndrome!
If you are running or supporting a rare epilepsy patient group, I recommend you also check these two additional resources to understand why these studies are so important and consider how it could work for your disorder:
1) The pre-competitive industry collaboration involving seven biopharmaceutical companies and the Loulou Foundation to run an observational study specifically designed to evaluate the feasibility and suitability of a collection of clinical outcome measures in CDKL5 deficiency: here and here.
2) The amazing Mike Granglia from SYNGAP Research Fund explains in a recent video why Natural History Studies specifically designed to inform on clinical trial design are so so important to not delay the initiation of clinical trials for these syndromes. Please watch it, the Natural History Studies part starts around minute 5:45 but the entire videos are fantastic.
2 – Gene therapies are coming for many epilepsies
There are many reasons to believe that many of the genetic epilepsies will benefit much from restoring gene expression in patients, even in older individuals. And we saw several studies in mice presented at AES where scientists returned gene expression to mice with genetic epilepsies showing very good efficacy. See for example this study in CDKL5 Deficiency Disorder, this one in SCN1A Dravet syndrome, and this one in STXBP1-related DEE.
To be able to do that in patients, scientists need to develop gene therapy approaches using antisense oligonucleotides (ASOs) or virus that can bring to the brain the missing gene, or virus that can bring to the gene CRISPR approaches or transcription factors (see review of options for Dravet syndrome here). We saw several of this presented at AES this year.
The most advanced program is the one from Stoke Therapeutics. In addition to presenting an update about their observational study, Stoke presented new information about how their ASO to increase SCN1A distributes within the brain in non-human primates, and the likely doses that are needed to produce enough sodium channel in the brain in trials. From their data, it seems that repeated administration of 30mg of STK-001 (their ASO to increase SCN1A levels) will be a good target to to achieve active brain levels in patients. They are not yet administering those levels to patients because the interventional trial is still at early stages, and it is a Phase 1/2 trial without any prior trial in healthy volunteers, therefore starting with only one administration of STK-001 to Dravet patients at very low dose and moving from there to higher doses, still with only one administration to monitor safety, and then starting with repeated dosing also from lower concentrations to higher. From this early data, Stoke reports good tolerability, and a trend towards having less seizures but all participants had received either only one administration of STK-001 or multiple administrations of 20mg, so we really have to wait for the trial to reach higher dose with repeated administration in order to see the therapeutic potential of this treatment. We might potentially see this first from their UK study, called ADMIRAL, where Stoke will evaluate multiple doses of up to 70mg [see Note for parents at the end of this text].
Behind STK-001, there are several therapies in development, all designed to increase SCN1A for Dravet syndrome, that have not yet started clinical trials. You can see a summary of all those approaches here, where I explain the biology of SCN1A deficiency and the options to increase it.
Encoded Therapeutics is developing ETX101, a gene therapy that uses an AAV virus to upregulate SCN1A in the brain. They are preparing to start clinical trials, and this year presented data on their observational study as well as on understanding caregiver perspective to better design their upcoming clinical trial. A company called CAMP4 has acquired the rights to the ASO program from OPKO to upregulate SCN1A, and talked about the preclinical data that is available for this program at AES. And there are at least three academic groups developing viral-based gene therapies for Dravet syndrome, all focused on increasing the SCN1A gene: the adenovirus program that we saw last year at AES, a new one this year, and a CRISPR-ON approach being developed in Italy.
We also saw preclinical data for a gene therapy program for SLC13a5 Deficiency, showing efficacy in young mice and also in young adults, which is very promising towards future studies. And scientists at UCL in the UK are working on bringing gene therapy to non-genetic epilepsy! Their gene therapy is designed to sense if neurons are hyperactive, turn on and produce potassium channels (which makes neurons less excitable), and as soon as neurons are not hyperactive anymore, turn off. This means that neurons would only make use of that gene therapy when they truly need it. They are still trying it in mice, so it is at early stages, but I found this research one of the most groundbreaking presented AES.
3 – Not all is gene therapy: Many exciting progresses with small molecule drugs
We had a very good year of progresses in small molecule drugs (“normal medicines”) for epilepsy, including for rare and non-rare epilepsies.
Probably the most impressive is the Phase 2 data of XEN1101 in adult patients with focal onset seizures, which is the most common seizure type. There are more than 20 drugs approved for treating focal onset seizures, and trials are done with the new drug added on top of whatever the patients are already taking, so it is very difficult to see impressive efficacy in this oversaturated space. Yet XEN1101 achieved a reduction in seizures of up to 52% at the highest dose. This is rare and exceptionally good.
There were also several updates on late-stage programs presented. We saw post-Phase 3 trial data for fenfluramine in Dravet syndrome, and for ganaxolone in CDKL5 Deficiency Disorder, both looking really good. Ganaxolone has also shown efficacy in PCDH19 epilepsy in a Phase 2 study, with 61,5% of seizure reduction. There is an ongoing trial in young children with diazepam nasal spray as a rescue medication, and Takeda presented how they plan to count seizures in Lennox-Gastaut syndrome in their Phase 3 trial with soticlestat which has a novel glutamatergic mechanism of action.
And I liked to see several innovative compounds coming up in the earlier pipeline. Eisai has a GAT1 inhibitor called E2730 as a potential alternative to tiagabine. A KCC2 activator might have potential for treating seizures in Rett syndrome. Praxis has a sodium channel blocker called PRAX-562 with preclinical efficacy for SCN8A and SCN2A Gain-of-Function epilepsies, and Xenon has sodium channel potentiators with preclinical efficacy for Dravet syndrome (SCN1A).
I might be missing some compounds, but it is clear that 2021 was a very good year for the developments of medications for epilepsy, with some really strong data in clinical trials and in the “real world”, and with new drugs that have novel mechanisms of action at early discovery stages.
4 – Understanding genetics in epilepsy
It is likely that most epilepsies have some genetic contribution. As we learn more about it, and genetic testing becomes more common, we will find many more individuals with rare genetic syndromes and start finding sub-groups of patients with “common” epilepsy that might respond better to certain treatments.
Xenon Pharma has a drug in Phase 3 for KCNQ2-related epilepsy, and they are one of the sponsors of the Invitae genetic testing program for epilepsy. So they interrogated the data from Invitae to see how many of the cases are due to KCNQ2 mutations. I knew this disease was one of the most common genetic epilepsies, but I did not expect the numbers that they got: KCNQ2 is by far the leading cause of neonatal epilepsy, and still the most common in children under 6 months of age. We urgently need early genetic testing for epilepsy to not miss these cases!
There are still new genetic causes of childhood epilepsy being found, like SLC7A3 and SYNJ1 mutations, and scientists are able to create mouse models of genetic epilepsies that require mutations in more than one gene. And a large collaboration of scientists has put together a portal called NDD-CNV to collect information on copy number variations in neurodevelopmental disorders (many with epilepsy) to assist with variant interpretation, which is very important for syndromes like Dup15q and 8p disorders.
And we saw several genotype/phenotype studies where scientists try to see if different types of mutations produce different prognosis which is a common question from parents after genetic diagnosis (see for example SCN1A and STXBP1). There is a little bit of correlation when you compare mutation type in hundreds of patients, but it doesn’t seem that we will be able to predict the development of a child just knowing the specific mutation that they have in a given gene. There was also a massive study in 11,500 patients with epilepsy that relied on phenotype keywords to see if they could be matched to genes, and it identified certain keywords that are preferentially associated with some syndromes like “hemiclonic” and Dravet syndrome, and that suggests that we could potentially use good phenotypic descriptions to prioritize genes to test in these patients. I still believe the best option is to just sequence all people with epilepsy for all epilepsy genes or the entire exome/genome, with particular urgency for young children.
5. Patient groups are building research-enabling platforms
This year we saw some very important research efforts led by patient groups, with the purpose of making it very easy for scientists and companies to work in their disorders:
The NDD-CNV portal is hosted by the Broad Institute and co-developed with the Dup15q Alliance, Project 8p, and Ring 14 US and Ring14 International. It is a natural synergy among structural variant disorders (as opposed to point mutations) and I encourage you to check out the link.
A similar effort, the GRIN Portal, is also hosted by the Broad Institute and developed in partnership with several national and international GRIN patient organizations, and provides information about the genes, the disorders that they cause when mutated and the consequences of specific variants to facilitate interpretation.
Yet another portal hosted by the Broad Institute is the SLC6A1 Portal, with analogous information about SLC6A1 and also with collaboration from patient organizations.
Shout out to Dr Dennis Lal from the Cleveland Clinic and the Broad Institute for making those three portals positive (and more, like the sodium channel portal), and being such an amazing advocate and promotor for patient-led research in genetic epilepsy.
The SYNGAP Research Fund and Ciitizen have also brought together the largest cohort of SYNGAP1-related disorder patients in a platform to collect real world evidence, which will help design clinical trials for this syndrome. And I already mentioned the SCN8A Natural History Study and STXBP1 Natural History Study which are also happening thanks to the effort and leadership of patient organizations.
I believe this type of effort is the best investment that patient groups can do, more than investing on any single research program. It requires that the patient group takes a leadership position in the field and builds the blocks that will facilitate and de-risk individual programs. And it has required a cultural change where clinicians and academic scientists are now very open to partnering with patient organizations and co-leading these efforts. 2021 was a great example of this.
LOOKING INTO 2022
As we look into the new year there are some planned milestones that I look forward to, and some things that I hope will also come true:
I look forward to efficacy data from STK-001 in Dravet syndrome, and the initiation of clinical trials with ETX101.
I look forward to ganaxolone getting approved for CDD, and fenfluramine showing positive results in the ongoing Phase 3 (if interim data is already available by then)
I look forward for more genetic therapies moving into trials. There are several preclinical programs with ASOs and viral-based gene therapies, and in the coming years we should see several of these moving into trials.
And I look forward to seeing progress with several small molecules that target disease-causing proteins, as in KCNQ2, SCN2A, SCN9A and SCN1A Dravet syndrome. Short of gene therapies these might be the best treatments for these disorders.
Ana Mingorance, PhD
Note for parents of someone with Dravet syndrome: when fenfluramine and CBD started trials in Dravet they already had data from adults with other diseases, same for soticlestat and others, so by the time they started trials in Dravet they could immediately administer the drug at the therapeutic dose daily for three months to see efficacy. That is different with STK-001 because it is a disease-specific treatment targeted to people with deficiency in SCN1A so they had to start trials directly in patients and figure out the tolerable and effective dose also in patients, going little by little and starting with low doses. The way I see it, it wouldn’t be correct to compare the current Phase 1/2 Stoke trial with a Phase 3 with other drugs. They are still building up to getting to the best doses, and so far so good.
Disclaimer: These are my own impressions from the presentations and topics that I was most interested in. I write these texts with the parents of individuals with rare epilepsies in mind, so excuse also my lack of technical accuracy in parts.

MAIN LESSONS FROM THE 2021 CDKL5 FORUM
For the past seven years the Loulou Foundation hosts an annual meeting where scientists and drug developers working on CDKL5 deficiency, together with representatives from patient organizations, meet to discuss the latest advances.
Here are the main news and take-home messages from the 2021 CDKL5 Forum that took place in November 1-2 2021.
For the past seven years, the Loulou Foundation has hosted an annual meeting, the CDKL5 Forum, where scientists and drug developers working on CDKL5 Deficiency Disorder (CDD), together with representatives from patient organizations, meet to discuss the latest advances in the field. You can find written summaries from the past three meetings here: 2018, 2019 and 2020.
The 2021 CDKL5 Forum edition took place November 1-2, and for the second year, due to the ongoing pandemic, it was held fully on-line. The Loulou Foundation turned this into an opportunity to also host a series of workshops prior to the 2-day conference, turning the Forum into a giant Think Tank. This made this year’s Forum quite unique, and I hope the pre-conference workshops are here to stay.
I know that many in the CDD patient community are waiting for this written summary, so I will try to extract the main conclusions from the Forum with this audience in mind. But I want to start with a warning: if you are reading this summary mainly looking for an answer to “when are the gene therapy trials for CDD starting?”, the best answer that we have today is “we don’t know yet”. I understand this might feel disappointing to some, as if we are not progressing fast enough, and potentially make you feel a bit hopeless. That’s only human, so I want to acknowledge that upfront.
In the next paragraphs I will walk you through the magnitude of the effort and the tangible progress that has taken place around research and treatments for CDD, and that was presented at the Forum. I invite you to read it in the spirit of celebrating each step and each progress and small victory along the way, because we have come a very long way, and this year the progress in biology understanding and therapy development has been particularly significant.
It is remarkable how far we’ve come in the last few years, and the size and commitment of the community of scientists and companies that has grown around CDD.
Because of that, I have structured the summary of this year’s Forum around the following themes: hope, tangible progresses, strategy and partnerships.
1. HOPE
The 2021 Forum started with the voice of the patient. This year it was IFCR’s President Karen Utley, who shared with the audience the life of her daughter Samantha, and the impact that CDD has on Samantha and her entire family. Samantha sees 14 different specialists a year and takes 15 different medications, which gives us some quantitative data for the complexity of this disorder. From the many great messages from Karen, I keep two: (i) small improvements can be life changing for patients and families, so please don’t focus only on cures and keep working to make more smiles and happy days, and to (ii) please keep hope alive for those who are feeling too overwhelmed to stay hopeful.
Her plead to keep hope alive was adopted as a motto by many of the conference speakers in the two days that followed.
There were several moments during the conference where science showed us there are many reasons for hope. These are some of those reasons:
Hope because mice show us that CDD is a good candidate for genetic rescue

Just two weeks before the Forum we celebrated the publication of a landmark study showing that CDD is reversible in mice (article link). Zaolan (Joe) Zhou, from the University of Pennsylvania, also presented the results of his study at the Forum, which show that CDKL5 is needed throughout life to keep proper neuronal functioning and that, in mice, turning “on” the CDKL5 gene in near-adult mice that grew up with CDKL5 Deficiency leads to large reversal of most symptoms. This is very valuable because it helps us learn what is biologically possible, and that CDD is not just a disease of neurodevelopment but also of neuronal maintenance.
In layman terms, it appears that in CDKL5 Deficiency, the brain developed fine and the problem is that neurons are not talking to each other correctly. So once we put the protein (or the gene) back they can start talking to each other correctly, because nothing was really lost: there was no neuronal death or failure to form those neurons, and they are not in the wrong location, which are all things that happen in other neurodevelopmental diseases. This study is a big reason for hope, and Joe received the CDKL5 Forum Award for Lab of the Year for this contribution to the field.
Hope because other diseases are already seeing great results
As part of the gene therapy session, Krystof Bankiewicz, from Ohio State University, showed us videos of children with AADC Deficiency who participated in a clinical trial for a gene therapy to deliver to their brains the AADC enzyme that they are missing. If you saw the videos you probably had chills. Patients with AADC Deficiency have very strong hypotonia, intellectual disability, dysautonomia and other complications due to missing an important enzyme necessary for producing dopamine and serotonin. Prior to treatment, the mobility of these patients is so impaired that they reminded me to several of the boys with CDD, unable to even hold their heads up. In the videos, we saw children go from that extreme hypotonia state to seeing changes 1 month after gene therapy, sitting after 6 months, walking with assistance of DAFOs after a year, and totally unrecognizable walking by themselves after 18 months. Krystof explained that many of the patients also became verbal, which was highly unexpected. While we don’t know yet how fast and how much people with CDD will improve once we have a gene therapy for them, seeing the results in this other enzyme deficiency help us learn what is biologically possible, and it is a big reason for hope.
Hope because science (and scientists!) keep coming up with great genetic tricks very quickly
While we are today very focused on gene therapies to bring an extra copy of CDKL5 to each neuron in the brain, we got to see at the Forum some of the science that is coming afterwards, and that will allow us to fix the mutated gene in patients with genetic diseases. David Liu, from the Broad Institute, showed us how he is using an advanced version of the CRISPR system to correct mutations where one letter is missing, and showed us how they can do it already in cultured cells from a girl with CDD (this is still all very early and being optimized). And Feng Zhang, also from the Broad Institute, showed us another advanced version of the CRISPR system that instead of correcting one letter in the DNA can swap large sections of a gene or an entire gene. So we can imagine one day getting a single gene therapy that gets to the brain and swaps the entire CDKL5 gene for a good copy, and that could potentially be used for kids with all types of mutations.
But before we get to see these gene editing approaches turned into actual treatments, we have many other therapies coming our away. I review those in our next section: tangible progress.
2. TANGIBLE PROGRESS
Progress in three gene therapies
Last year at the 2020 Forum we talked about the two gene therapies for CDD that had shown to have efficacy in mice deficient in CDKL5, or as we call it in science had “preclinical Proof-of-Concept”. We also talked about how gene therapies need to go through three stages: first prove that you can treat a mouse, then prove that you can use it in larger brains (non-human primates) since it is much more difficult than in small mouse brains, and then finally start the first trials in a very small group of patients to determine safety before exposing more patients.
This year we saw not two, but three gene therapies, and how the most advanced ones are now working on that second stage that goes between showing that you can treat mice and being able to start clinical trials.
The gene therapy that we saw presented for the first time is the program between the University of Pennsylvania (Penn) and the company Amicus, and it is a very interesting hybrid between gene therapy and protein (enzyme replacement) therapy. They call the approach cross-correction, because the therapy uses a virus to bring to neurons a copy of the CDKL5 gene (this is a gene therapy), with a special version of CDKL5 that will produce a secreted protein. The idea is that the neurons that are successfully corrected will act as local sources of enzyme to supply to neighboring cells. This was the first time that we saw mouse data from this project at the Forum, and the researchers are trying to see how much more efficacy they can get by using this special version of CDKL5 as opposed to plain CDKL5.
For the other two gene therapies, from Penn (in collaboration with Elaaj Bio) and Ultragenyx, we got to see some of the work that goes into getting the therapies ready for patients now that we know both of them work in mice.
The gene therapy program from Penn/Elaaj came with new EEG data, showing that it is not only the symptoms but also the EEG abnormalities in mice with CDKL5 deficiency that get corrected with the gene therapy. And more importantly, they showed two newer versions of the gene therapy, more advanced than the one we saw last year, and how with each iteration they are getting better and better distribution in the non-human primate brains. They are making these upgrades trying to get as much expression of CDKL5 and as broad as possible, and as the presenter said, “we are getting closer to the target of robust expression in the brain” that they want to see before moving into clinical trials.
The team from Ultragenyx also dedicated most of their presentation to talk about those next steps after seeing efficacy in mice with CDD (which we saw last year). They told us about the need to fully evaluate safety and toxicology in animals before entering the clinic, and also about all of the additional work that they are doing to prepare for clinical trials involving natural history and biomarker data. This was very impressive, and showed how Ultragenyx has many people in the clinical and regulatory teams and many other teams in addition to their preclinical researchers all working on the CDKL5 program.
So we don’t know yet when trials might start, but we know that these three programs keep getting closer and closer to the clinic, and that we have some of the best people in medicine working on solving all of the difficult barriers that need to be overcome to take these treatments into clinical trials (like better brain distribution, and development of biomarkers, and clarity on clinical scales to use). You will see more of this in the last section about strategy and partnerships.
Progress with ganaxolone
If everything goes well, ganaxolone will become the first drug to be approved for the treatment of CDD. The FDA plans to make a decision in March 2022, and the EMA some months later. In the meantime, Marinus has an Expanded Access Program ongoing which is now also open to Europe.
What I found really exciting from the Marinus presentation was data from their open-label studies, where they showed how patients from the Phase 3 trials are doing as they rolled over into the open-label extension. Since it is also openly available in their website and not confidential, I am going to copy their slide here because the data looks really good and it is better seen in the graph:

In the solid green bars, you see the patients that received ganaxolone during the Phase 3 study, and then continued taking it in the open-label extension. Remember that the study is blind, so no one knows if they are taking placebo or ganaxolone during the 17-week study. These patients had a seizure reduction of 30% at the end of the trial, and for the following 12 months their seizure control got better, staying more around 40-45% seizure reduction. This is not trivial, because it is common for patients with CDD to respond to a new medication for 3 or 4 months and then lose efficacy, while ganaxolone doesn’t show signs of losing any efficacy after a year, all the contrary.
And in the striped green bars you see the patients that received placebo during the Phase 3 study, but then started taking it in the open-label extension. Because the study is blind no one knew during the 17-week study if they were getting the drug and not responding to it or if they were in placebo. On average this group had seizure reduction of only 6,9% during the trial (since they were not on the drug), and as they rolled over into the open-label extension their seizure control got better and better until they caught up with the ones that we already taking ganaxolone in the trial.
This is a beautiful graph, that shows that ganaxolone has better efficacy in CDD than what is seen only during the double-blind phase, and that you have to give it enough time to show its full potential. It is also quite encouraging to see how stable the efficacy of ganaxolone remains over time.
Progress with fenfluramine
Fenfluramine is an anti-seizure drug that is already approved for Dravet syndrome where it has exceptionally high efficacy in reducing seizures. After a successful small pilot study in CDD run at NYU, the company Zogenix has been working on preparing for a Phase 3 trial of fenfluramine in CDD.

And the Zogenix team gave us the great news at the Forum that the Phase 3 trial with fenfluramine in CDD has already started and is already enrolling patients in the US clinical sites! They are enrolling children and adults with CDD as young as 2 years old, and once they have enough safety data they might open it to 1-year-olds and older, and patients need to have active epilepsy since that is what the study is measuring. For the trial, Zogenix is looking for 100 participants.
This is really good news because it means that at least 100 patients with CDD will get to try fenfluramine, and if it has in them even a fraction of the efficacy that we have seen in Dravet syndrome and in the small study in CDD, it will be very meaningful for those patients and their families.
3. STRATEGY AND PARTNERSHIPS
The Forum becomes a Think Tank
During the week prior to the conference, the Loulou Foundation hosted focused workshops that covered four areas of priority for the development of therapies for CDD:
Spontaneous and induced seizure activity in CDKL5 knockout animal models
New technologies for improved therapeutic cargo delivery in the central nervous system
Biobanking of patient samples and fluid biomarker discovery/validation to support CDD therapeutic development
Functional biomarker development/validation and inclusion in clinical development plans
For each workshop, the Foundation invited a group of academic, industry and clinical researchers, and for some also patient representatives, to try to map out (i) where we are, (ii) where we need to be, (iii) what are the technologies or fields that we should be paying attention to, and (iv) how we can get organized to address those challenges including specific proposals for collaborations. This is what turned the 2021 Forum edition into a true Think Tank, and the outcomes from each of the workshops were presented during the main conference to share with the entire community.
To give you an example for how these workshops worked, when talking about new technologies for delivering genes to the brain, we covered all approaches from engineered viruses to synthetic biology, nanoparticles and the possibility to attach CDKL5 to proteins that cross the blood-brain-barrier to use them as shuttles. And we had scientists from companies that are developing all of these approaches sit on the same workshop and share with everyone else their thoughts and proposals. This level of collaboration among traditional competitors is highly unusual, and it is one of the themes that we kept seeing during the conference: the building of a scientific community around CDD that includes many companies joining forces to help us get to better treatments. You will hear more of this later, when I talk about the CANDID study.
The long road to solving the translational puzzle
Translational medicine refers to the bridge between treating mice and treating people. It seeks to answer questions such as: “how will we know that we have increased CDKL5 protein in the brain?”, “how will we know if we are improving synaptic function?” and “how are we going to measure improvements in patients in the trial?”.
To answer these questions, we need to develop ways to see things, which can be biomarkers (like things you can see in a blood test, or brain scan) or clinical scales. Because CDD is a relatively newly-described disease, we still don’t have these biomarkers and scales.
The good news is that there are many collaborative efforts ongoing to address all these questions and develop all these biomarkers and outcome measures. This was a central theme of this year’s Forum and the subject of two of the pre-meeting workshops, with important presentations on these topics:
Need and plans for a biobank of patient samples, with standardization of analytical techniques and data sharing including universities and companies
EEG-based biomarkers, presented by Eric Marsh from CHOP, as a way to measure dysfunction of brain activity in mice and patients with CDD, to detect potential restoration with treatments
Blood-based biomarkers, presented by Max Bianchi, from the Trinity College Dublin, as another way to measure dysfunction of brain activity in mice and patients with CDD, to detect potential restoration with treatments
A US and Australia NIH-funded study coordinated by Tim Benke from the University of Colorado to also test and validate biomarkers as well as new clinical scales for CDD in an observational study
An international observational study coordinated by the Loulou Foundation in partnership with 7 pharma companies to test and validate clinical scales for CDD, called the CANDID study (see also next section)
In addition to helping us solve the translational puzzle, what all of these efforts and studies have in common is that they all require the active participation of the patient community. This could look like organizing the collection of some blood samples at your next national families meeting to then send them to Dublin, or participating in an EEG study in Philadelphia or Boston, or volunteering to participate in the observational studies that will take place in clinics from around the world. You will likely hear of some study near you!
So if you have a family member with CDD, I encourage you to consider participating in some of the studies as well as enrolling into the CDKL5 Registry (this one only needs data, no samples or visits to a clinic needed).
We cannot answer all of these key translational questions without you.
A first-of-its-kind partnership
Just a week before the Forum, the Loulou Foundation announced the launch of a pre-competitive industry collaboration involving seven biopharmaceutical companies together with the Loulou Foundation. The consortium will run an observational study specifically designed to evaluate the feasibility and suitability of a collection of clinical outcome measures in people with CDD from all ages.
Xavier Liogier, from the Loulou Foundation, explained how this study fits into a larger roadmap for preparing the CDD field for the arrival of better therapies and cures, and that already started with the PFDD meeting in 2019 and getting an ICD-10 code for CDD.
Observational studies are like a clinical trial but instead of testing a drug you are testing the clinical scales to check that they are good for that disease. These studies are very important to be ready for clinical trials that look at several symptoms or disease domains. Usually, when companies run observational studies of this kind, they do it alone, so in diseases where there are many companies developing therapies we might end up seeing several observational studies, each one run by a different company with a slightly different design.
We wanted to avoid this for CDD.
The idea for the CANDID study was born two years ago as a big hairy audacious goal: what if we could get all of the companies working on CDD come together, and co-design a single observational study that would meet everyone’s needs, get also the FDA to review it while we are at it, and then run that single study with all of the companies working together to minimize the number of observational studies that we need in CDD.
It sounded good. And all of the clinical teams at the different companies that we talked to also told us that it would be something they would support. That’s what gave us the conviction that we needed to try to make it happen. The problem was that we had no reference for other collaborations of this type. There was no book that we could read to know how to do this. This was going to be a first-of-its-kind partnership.
So for the past two years we had to figure out a way to co-design a study among so many parties without the study dying of decision paralysis, and we had to figure out the best way to build a consortium to govern the study where all companies would be equal partners. We also had to grow as a Foundation to be able to lead and coordinate these efforts, which brought us new team members like Xavier Liogier, who has the ideal human and professional mix to lead this study, and new strategic partners in clinical operations to help run this observational study with the very same standards as a pharma-run Phase 3 trial.

This is why I am so proud to be part of this story, as part of the Loulou Foundation team that made the CANDID study possible. We knew that all the companies working on CDD would like to see a consortium-run study, and we all understood that we needed to try to avoid having multiple corporate observational studies in CDD. So we figured out how to make this first-of-its-kind partnership happen, and in the process, wrote the first chapter of that book.
The CANDID study is about to start and will look for 100 participants of all ages (with active epilepsy or not), hoping to include study sites in the US, Canada, UK, France, Spain, Italy, Germany and Russia. This will help us make the study accessible to the large majority of patients diagnosed with CDD.
Strategy and partnership also on the advocacy side
Natalie Ladly, secretary for the International CDKL5 Alliance, helped close the conference with a presentation about the Alliance and a call for action.
There are 17 patient organizations under the Alliance (check them out at https://cdkl5alliance.org/) and one of the top priorities is to help the community be trial-ready. Natalie explained that they have 47 clinical trial sites around the world, at least one on each of the countries under the Alliance, and made a call to the industry and the medical community to reach out to the national patient groups and the Alliance and tell them what would help them (industry and clinicians) the most. Essentially, “help us help you”, and “by the way we have solved your problem for finding trial sites”.
I believe every year I write a longer summary. There is just so much going on, and so many news and progresses in many areas, that it is difficult to know what to cut from the summary. My main impressions from the 2021 CDKL5 Forum are that we are seeing solid tangible progresses in many areas, and that we seem to be turning a corner, where the ratio of individual labs working on CDD versus partnerships is changing and now a majority is working on the same agenda to solve the same problems together. This partnership model was started by the academic and clinical centers, and it has now also reached biotech and pharma companies.
This is why I want to commend in particular the professionals from so many biotech and pharma companies (we had 24 companies at the Forum!) that came to the Forum and sat down with their traditional competitors to share their best ideas for CDD and agree on joint efforts to make these happen. And I believe that the industry consortium behind the CANDID study is a testimony to that spirit of breaking previous barriers to industry collaboration, not just by saying nice words, but by working together to make progress actually happen.
And that is why hope, tangible progresses, strategy, and partnerships, where the themes of the 2021 CDKL5 Forum.
I hope you enjoyed this summary. Please let me know your thoughts in the comments.
Ana Mingorance, PhD
Disclaimer: These are my own impressions from the presentations and topics that I was most interested in as a scientist and patient advocate, and not an official text about the Forum by the Loulou Foundation. I write these texts with the parents of individuals with CDD in mind, so excuse also my lack of technical accuracy in parts.

CDKL5 DEFICIENCY IS REVERSIBLE
CDKL5 deficiency is biologically reversible, bringing much hope and creating a sense of urgency for the development of restorative treatments for CDKL5 deficiency disorder (CDD). These are the conclusions of a publication from Zhaoland (Joe) Zhou’s lab from the University of Pennsylvania. The study was just published in the Journal of Clinical Investigation, and it is poised to become a landmark study in the field.
CDKL5 deficiency is biologically reversible, bringing much hope and creating a sense of urgency for the development of restorative treatments for CDKL5 deficiency disorder (CDD).

These are the conclusions of a publication from Zhaolan (Joe) Zhou’s lab from the University of Pennsylvania. The study was just published in the Journal of Clinical Investigation, and it is poised to become a landmark study in the field.
It has been almost 15 years since a similar publication from Adrian Bird in mice with Rett syndrome due to mutations in MECP2. Bird’s lab showed that Rett syndrome was reversible if the gene was returned to adult mice who had developed without MECP2. The study told us that it is likely never too late to rescue MECP2 deficiency, and it encouraged research into the genetic rescue of Rett syndrome in patients. The experiment (and the investigator) became so famous, that gene therapy researchers from universities and industry have been asking for the last 15 years “has anyone already done the Adrian Bird experiment for this gene?” for any genetic syndrome that they consider working on.
Joe Zhou and his colleagues just did that for CDKL5. And the response is affirmative: CDKL5 deficiency is biologically reversible.
Here is a summary of these findings, and what they mean in the context of developing curative treatments for CDD. There is also a press release from Penn here.
WHAT THE PUBLICATION SHOWS, IN LAYMAN’S TERMS
I love the elegance of this study, and I am impressed by the amount of work and exquisite diligence that went into creating this publication. This is why I feel so strongly about the importance of these results.
Terzic et al (2021) creates two types of genetically modified mice. One type is mice that are born with normal CDKL5 expression, but that can stop expressing CDKL5 when the investigator decides. The other type are born with CDKL5 deficiency, but can produce CDKL5 when the investigator decides. Essentially there is one mouse strain where scientists can decide when the disease will start, and one where scientists can decide when it will stop. And they use these mice to answer key questions about the biology behind CDKL5 deficiency.
Because the protein CDKL5 is expressed in the brain throughout life, the scientists hypothesized that perhaps it is important also throughout life, not just in early development. This is not trivial. People born with a bad copy of CDKL5 develop a disease called CDKL5 deficiency disorder (CDD) that starts very very early in life, leading to global developmental delay in addition to seizures and other complications. Because it starts so early, it looks like having enough CDKL5 is important for very early development, and maybe if you make it past all those critical early stages of post-natal development with normal expression of CDKL5 then you will always be ok even if you stop producing enough CDKL5 later in life. Short answer: you are not.
It is never too late to develop CDD. That is why the protein CDKL5 is expressed in the brain throughout life, because it is always needed.

This is what the publication shows, by letting mice develop normally and then turning off their CDKL5 gene when they are young adults. By that age, mice that are born with CDKL5 deficiency already have all their symptoms, which include cognitive, motor and behavioral deficits, as well as alterations of neuronal excitability and synapses. Same as for people with CDD. And the first important finding of the publication is that if you let mice grow up to that age completely normal, and then turn off their CDKL5 gene, they will progressively develop almost all the same symptoms of the disease (except for less motor symptoms). Pretty much as if they had been born with CDKL5 deficiency.
And that is the first conclusion: CDKL5 deficiency looks like a disease of neuronal maintenance, not so much of neuronal development. We talked about that at the 2020 CDKL5 Forum (read it here).
The second part of the study gets better: if CDKL5 is so important for the mature brain, what happens if we bring it back at that point? How much can we get back?
For this the Penn team uses the second type of mice, the ones in which they can stop CDKL5 deficiency at any age they want. And they let them grow through all those key developmental ages with CDKL5 deficiency, and they let them display all of the disease symptoms. And then, when mice are early adults, they turned on the gene so they no longer lacked CDKL5. This is the mouse version of what would happen if we already had the perfect gene therapy for CDD and we treated patients that are no longer children.
And the results are beautiful and make this study a landmark in the development of curative treatments for CDD. Barbara Terzic and Joe and their colleagues show how later-age restoration of CDKL5 expression rescues most CDD deficits. They re-start CDKL5 expression in mice that are of young adult age, that already have developed all their CDD symptoms, and then see reduction or full rescue in many motor, cognitive and behavioral readouts. Mice with CDD don’t develop easy-to-measure seizures, which is a difference with how the disease presents in people, but the Penn group was able to interrogate brain circuit hyperexcitability using an excitatory neurotransmitter similar to glutamate to “poke” neurons, and saw how the hyperexcitability that comes with CDKL5 deficiency was also rescued by reintroduction of CDKL5 expression. They also used this neurotransmitter to induce seizures in the CDKL5 deficient mice since they are prone to epilepsy, and saw how this was also rescued by reintroduction of CDKL5 expression. They even looked at the presence of receptors in synapses, which is altered by CDKL5 deficiency, and this was also restored in the adult-rescued mice. So I want to highlight again how thorough and well-done this study is: it is very clear from this study that reintroducing CDKL5 expression later in life is able to correct functional and behavioral deficits across the board, not just a few symptoms.
And that is the big beautiful conclusion of the study: the promise of disease reversal in CDD, across a broader-than-expected time window.
WAS THIS NOT OBVIOUS OR KNOWN BEFORE?
No it wasn’t.

During brain development neurons need to be born, then need to migrate to where they are needed in the brain, grow connections to the parts of the brain that they need to be connected to, and then do their job properly remaining in place until we get old and neurons start dying. In many neurodevelopmental diseases neurons might fail to develop properly, or might migrate to the wrong places, or not connect with who they should, or suffer from neuronal loss. And for all those it is too late to come with the gene after those problems have happened.
We knew that CDD looked quite good for a potential genetic rescue in older individuals. We knew the neurons where there, in the right location, that brain connections looked good, and there were no signs of neurodegeneration. At the biological level CDD looks more like a channelopathy, like Dravet syndrome (Nav1.1 channel), than a proper developmental disease.
But from thinking “the odds are good” to being able to say “at least in mice this disease is reversible” there is a long stretch. That is why this study is such a landmark.
The results from the Adrian Bird study in Rett syndrome are actually an exception. Similar studies in related diseases have failed to show such an across-the-board rescue. For example in Phelan-McDermid mice, reintroduction of the SHANK3 gene in adult mice only rescues some symptoms, and early postnatal intervention is needed for across-the-board rescue (link). And in the Angelman mice (Ube3a gene), you can only rescue many of the symptoms if you restore the gene during early development, and the authors concluded that “adult reactivation of Ube3a is only minimally efficacious as a therapeutic intervention” (link).
So with CDD starting so early in patients, and with the clinical symptoms being so severe, it was not obvious at all that CDD would look so promising for rescue in older individuals and not more similar to the mouse study for Angelman.
WHAT IT MEANS FOR GENE THERAPIES
This study means that we now know that CDKL5 deficiency is biologically reversible. Now we need to get the tools to do that in the clinic.
What these genetic rescue studies in mice have in common is that they use mice that are genetically re-programmable, where we as scientists can choose to turn on or off a particular gene defect. People is not born like that, we cannot choose to turn off their genetic disease. We need therapies for that. These could be therapies like gene therapy, where we bring a new copy of the CDKL5 gene to their neurons, or other therapies like gene editing that fixes the mutation that they have, or even protein therapies if we can supply enough CDKL5 protein to their brain.
So there are many technical possibilities to restore CDKL5 in patients, but there are also many challenges to do this. This is why developing this type of treatment takes so long. One of the main challenges is getting to all (or most) neurons in the brain. CDD is not a disorder that impacts a small brain region, and where we only need to fix it there with an injection. It actually involves the entire brain, and we would need to make sure all (or many) of those neurons have sufficient CDKL5 as a result of a treatment. This sounds simple but is tremendously difficult. Understanding how many neurons need to be rescued to have enough efficacy, and how much CDKL5 exactly do they need (is it ok with half of the amount?), and having the right therapy that can do that in a safe way… it is not easy.
But the good news is that this is a challenge for ALL neurological diseases, so scientists and companies around the world are trying to make sure we know how to make treatments that can put back a gene (or fix it) throughout the brain. This will benefit CDD and many others. And it is also good news that we already have several of these scientists and companies working on developing these treatments for CDD, and we have already seen some early results with two of these gene therapies in mice with CDD (see here).
It will only get better from here. I expect this study to attract more attention, more research and more resources, to developing curative treatments for CDD now that we know biology is on our side.
In summary:
The part of the puzzle that we could do nothing about, whether the brain has a narrow temporal window where it needs CDKL5 or not, is the one where we got very lucky. CDKL5 deficiency is biologically reversible.
The second part of the puzzle is to build those therapies, and that one is not about luck, but about science and time and money. We can do much about it.
This is like the difficulty of building a rocket to go to the moon: it is hard, but you know that your destination is waiting at the other side.
Now that we know that moon exists, that the disease is biologically reversible, all hands on deck to build that rocket.
PS1: Thank you to the Penn teams for working so many years in this study. This study had financial support from the Loulou Foundation, IFCR and several NIH grants. Science is not easy or cheap. And thank you for making the publication open access!
PS2: The 2021 edition of the CDKL5 Forum is coming up in November 1-2, and this study is only one of the several good news that will be presented at the meeting.
Ana Mingorance, PhD
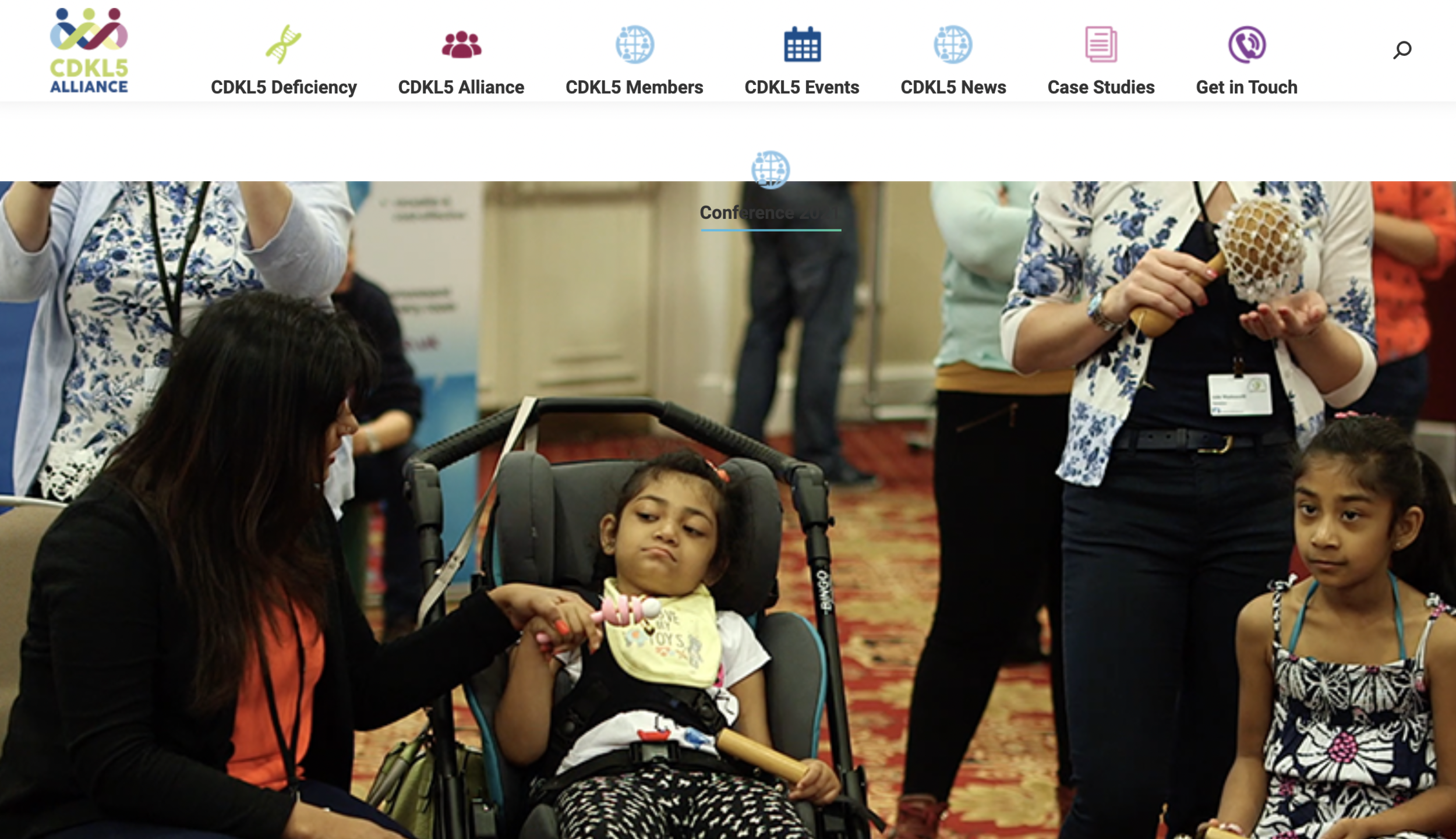
2021 CDKL5 ALLIANCE Virtual Family Conference
Last weekend, the CDKL5 deficiency disorder (CDD) community gathered in front of their computers for a virtual annual meeting organized by the International CDKL5 Alliance. For the 2021 Alliance meeting, the organizing team commission a series of pre-recorded videos to a large range of speakers and published the videos with subtitles so that all CDD families from all around the world could watch them at the same time. You can also access the videos at the CDKL5 Alliance website. Here is a personal summary of the 2021 CDKL5 Alliance meeting.
I miss conferences in person a lot. And the type of conference in person that I miss the most is the annual patient conferences organized by the rare epilepsy patient organizations that I collaborate with.
Last weekend, the CDKL5 deficiency disorder (CDD) community gathered in front of their computers for a virtual annual meeting organized by the International CDKL5 Alliance. Did you know that there are about 20 countries in the Alliance? That is how large and connected this community is. The last face-to-face meeting took place two years ago and I strongly encourage you to read the summary from that meeting.
For the 2021 Alliance meeting, the organizing team commissioned a series of pre-recorded videos to a large range of speakers and then published the videos with subtitles so that all CDD families from all around the world could watch them at the same time. You can also access the videos at the CDKL5 Alliance website.
Some parents of children with CDD have asked me to write a summary of the virtual conference, since a written summary is always easier to translate and digest. For all of you here is a personal summary of the 2021 CDKL5 Alliance meeting, sprinkled with some opinions as I often do.
1. THE SCIENCE: HIGHWAYS AND HEADQUARTERS
Prof John Rouse from Dundee reviewed where we are in our understanding of what CDKL5 does. In the recent years we have made large progresses on our understanding of what CDKL5 does in the cell and therefore what goes wrong when CDKL5 is missing.
Today we know that the protein CDKL5 turns other proteins on and off as if they had a light switch, and many of these proteins that are controlled by CDKL5 are associated with the neuronal skeleton (the cytoskeleton). The neuronal skeleton is not like our bones, which are essentially there to give us some structure. In cells, the skeleton is more like highways that are used to transport things to where they need to be, for example making sure that glutamate receptors get to the right spot in the surface of the neuron for the neuron-to-neuron connection to work fine.

And in addition to being important for the highways, it turns out that CDKL5 has a second job at the cell nucleus, which is basically the headquarters or control center in the cell from where all decisions are made. But what could CDK5 be doing there? This is what keeps John awake at night, so he has put together a team of scientists to try to answer this question. It turns out that in the nucleus, where all of the copies of all of our genes are located, the genes (made up of DNA) are constantly suffering small damages that the cell quickly repairs so that all genes work properly. And CDKL5 is a member of that DNA damage surveillance team. It does that by turning on other proteins that are needed to do the repair. And it also helps the cell read some of these genes. So CDKL5 is really important for the nucleus to function properly.
With the very complex (and multiple!) roles of CDKL5 in the cell, it is clear to me that the best way to fix the disease is to put the gene or the protein back, or to fix the mutation. Anything that will produce functional CDKL5 protein again. Trying to bypass a protein that is missing is doable if the function was only one, but for CDKL5 there are too many functions to try to fix, so I would focus on bringing CDKL5 back.
2. CLINICAL TRIALS: WHERE ARE WE TODAY
Before a drug or a gene therapy can be approved to be used to treat patients it has to progress through clinical trials. I think this year with the coronavirus vaccines we are all very familiar with how clinical trials work, so I will only explain that in rare diseases like CDD we have two stages of clinical trials: Phase 2 trials, where a small group of patients get the treatment, and Phase 3 trials, where many more patients from many countries get the treatment and usually half of them get placebo (but are offered the treatment at the end of the study anyways). If the Phase 3 is successful, then the company can apply for marketing authorisation. The entire process for trials and regulatory review for approval ends up taking 4 to 5 years.
We still don’t have gene or protein therapies for CDD, so while we wait for scientists to develop them we are trying to discover medicines that could treat the symptoms of CDD, helping all patients have a better life until we get to the curative treatments. We have had some clinical trials already with symptomatic treatments in CDD.
Last year we had very good news about clinical trials: the first Phase 3 trial in CDD, ganaxolone from Marinus, had been successful.
This year we also have very good news about clinical trials: we are about to start our second ever Phase 3 clinical trial in CDD, this time the drug is fenfluramine from Zogenix.
At the Alliance conference, Marinus reviewed their good data with ganaxolone in CDD. They are offering ganaxolone to all of the trial participants that want to keep taking it, and in the USA they are also offering it to other families who want to try ganaxolone before it gets approved. This is done through an Expanded Access Program. Ganaxolone works by acting on GABA, the inhibitory signal in the brain. If everything goes well ganaxolone will be the first drug to be approved for CDD.
Last year there were two Phase 2 studies in CDD. One was with the drug soticlestat, from Ovid and Takeda. This drug works by acting on the glutamate system, which is the excitatory signal in the brain. The companies were testing the drug in four different epilepsy syndromes, and after reviewing their data Takeda has announced that they will run Phase 3 studies in Dravet syndrome and Lennox-Gastaut syndrome. They still have not announced what they will do with CDD, and they did not present at the conference so we will have to keep an eye on any news about this program.
The second Phase 2 study from last year was with fenfluramine, which works by acting on the serotonin system, a modulatory signal in the brain. Fenfluramine has already been approved in the US and Europe for the treatment of Dravet syndrome, where it has impressive efficacy to reduce seizure frequency. Zogenix presented an overall introduction to the company at the Alliance meeting and didn’t talk much about their plans for CDD, but they have already announced that they are starting a Phase 3 trial in CDD with fenfluramine starting this year. Phase 3 trials are international, happening in many countries, so I look forward to the announcement of which countries will be included in this study. And if the Phase 3 results look like the small Phase 2 study results, we could have potentially a second drug approved for the symptomatic management of CDD.
This progression of clinical trials for CDD is exceptional! Only 5% of rare diseases have any drug approved, which means that 95% have nothing. And we are having these two Phase 3 trials back to back in CDD, so we are on track to join that 5%!
But these symptomatic treatments are only the tip of the iceberg. Right below we have a growing pipeline of therapies in development that will correct the lack of CDKL5.

3. THE FUTURE: FROM SYMPTOMS TO CURES
If you have a child with CDD the main answer that you probably wanted to get from this conference is WHEN are gene therapies starting clinical trials.
And I am sorry to say that the answer is “we don’t know yet”. Because we cannot know yet, nobody does. There were four different groups that presented data at the Alliance meeting showing their progresses towards developing a gene therapy for CDD. That is four different shots on goal, four independent efforts like when many companies all started developing vaccines for COVID without knowing how many would succeed and how fast it would happen. And we are following the same approach in CDD: many efforts all at the same time going as fast as they can.
The university of Bologna is developing a mix between an enzyme replacement therapy and a gene therapy. An enzyme replacement therapy is “protein therapy”, a type of treatment where you add the brain the protein that is missing, in our case the protein CDKL5 already made. For a gene therapy scientists use a virus to bring to cells a copy of the CDKL5. It is similar to the AstraZeneca or the Janssen vaccine but instead of using a virus to carry to the body the sequence of the coronavirus spike we use them to carry the sequence for the CDKL5 gene. The university of Bologna is using a virus to deliver to neurons the sequence for the CDKL5 gene, but it has been tweaked so that the protein CDKL5 will then be secreted by those neurons to all the surrounding ones, doing “local enzyme replacement therapy” on top of gene therapy. This is also called “cross-correction”. It is a cool idea. Amicus is doing something very very similar, in addition to classical enzyme replacement therapy with the purified protein. And then Ultragenyx and the University of Pennsylvania are doing the classical gene therapy using a virus to carry the sequence for the CDKL5 gene to the neurons in CDD. That means that we got to hear about FIVE different gene therapy or enzyme replacement approaches at the patient conference, by four groups since Amicus has two different programs. That is a lot.
The university of Bologna showed data using their cross-correction therapy in laboratory cells and mice. This is still early work at a university so it is too early to know how much work the experimental treatment still needs before it could move to trials.
Amicus presented some data about their cross-correction therapy program and their classical enzyme replacement therapy program. It was not an in-depth project review, but more of an overview of all the work that Amicus is doing in CDD which includes not only these two programs but also helping understand better how CDKL5 works and developing biomarkers (detection of special signals using EEG) that will help check if therapies are working in CDD mice and patients much faster. CDD is clearly important to Amicus. They are doing many experiments in mice with both treatments to see how well they could prevent or reverse the symptom of the disease in mice with CDKL5 deficiency.
The University of Pennsylvania presented a lot of data in mice with CDKL5 deficiency using a gene therapy to show that it is possible to prevent most of the symptoms in CDD mice. I say “prevent” because they give the gene therapy to mice when they are babies. I really liked the presentation, because they showed how the gene therapy was effective in many disease symptoms. That is the main different with symptomatic treatments: with gene therapy, you put the gene back and you are treating all of the different symptoms at once by removing the cause of all of them. This group has also run initial experiments in larger animals (macaque monkeys) to check safety and because we need to check that the gene therapy can get to enough neurons in larger brains, which is literally a much larger challenge than delivering the gene therapy to the brain of a mouse which is the size of your thumb nail. They explained that they are currently optimizing the gene therapy so that then they can go to trials.
The last presentation was from Ultragenyx, who are also developing a classical gene therapy similar to the one from the University of Pennsylvania. I also liked this presentation very much because they treated CDD mice at older ages, the equivalent of children 4-11 years of age, so what they showed to us was that it is possible to reverse many of the symptoms in CDD mice. They saw reduction of cognitive, motor and behavioral symptoms in the mice, and even used the gene therapy to correct neuronal connection problems in “human neurons” which were produced from patient skin biopsies and turned into neurons in the lab. Just like for the University of Pennsylvania, they explained that they are currently optimizing the gene therapy to be able to get into many neurons in larger brains (monkey) so that then they can go to trials.
As soon as one of the groups considers that their gene therapy is optimized enough, they will start the process to request a clinical trial permission and at that point we will finally get a date for when clinical trials might start. I hope you all realize how much work all of this is, and how lucky we are to have these five protein and gene therapy programs being all developed at the same time for CDD!
4. DATA, DATA, DATA
There were several presentations about why the international CDKL5 patient registry is so important, the international CDKL5 patient database from Australia, a study to validate several severity and symptom assessment for CDD, and an international observational study that is about to start and that is coordinated by us at the Loulou Foundation.
At the core of all of these efforts is a common theme: we need to understand better the symptoms of CDD and how they change over time (the natural history), we need to identify good scales to measure these symptoms, and this information and scales are absolutely essential to be able to tell if a treatment is working beyond just reducing seizures.
For the registry and the database, you can provide data about your child from your living room, and for the observational study we will try to get as close to your town and living room as possible!
By the way, have you noticed that clinical trials all have names? For example the trial with ganaxolone in CDD is the Marigold study, and the famous trials with spinraza in SMA was called Nurture. The Loulou Foundation is requesting suggestions from the community to name the international observational study so please reach out with your proposed names! (you can get some ideas here)
5. THE COMMUNITY
Although we didn’t get to meet in person, we did get to see many of the visible faces of the CDD patient community at this virtual conference.
Patric Benz from the Swiss CDKL5 patient group was the meeting host on behalf of the German-speaking national patient associations who were the organizers. His greeting in several languages and the fact that the videos were available with multiple language subtitles was a testimony to the reality of rare disease patient communities: at the same time dispersed and united.
During the conference we also saw images of people with CDD kayaking, skiing, ice skating and cycling. If there is something notable about this community it also their fearlessness and determination.
To conclude the conference, Nathalie Ladly from CDKL5 Canada and Antonino Caridi from CDKL5 Italy spoke about some of the big efforts of the Alliance in this last year, like developing the organization manifesto and sourcing a list of all possible clinical trial sites world-wide recommended by the community so that we can accelerate clinical trials. The goal of the Alliance is to expedite treatments and a cure for CDKL5, so of course a global pandemic would not stop them from putting together such a great conference program.
Next stop is the CDKL5 Forum in November, also full of updates about the treatment programs in development for CDD. Hopefully by then we get to meet in person.
I hope you enjoyed this summary! let me know your thoughts in the comments.
Ana Mingorance, PhD
Disclaimer: These are my own impressions from the presentations that I was most interested in, and not an official text about the Alliance meeting by Alliance or the Loulou Foundation. I write these texts with the parents of people with CDD in mind, so excuse also my lack of technical accuracy in parts ;-)